the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 Jun 2020
| 03 Jun 2020
Database for the kinetics of the gas-phase atmospheric reactions of organic compounds
Max R. McGillen
William P. L. Carter
Abdelwahid Mellouki
John J. Orlando
Bénédicte Picquet-Varrault
Timothy J. Wallington
We present a digital, freely available, searchable, and evaluated compilation of rate coefficients for the gas-phase reactions of organic compounds with OH, Cl, and NO3 radicals and with O3. Although other compilations of many of these data exist, many are out of date, most have limited scope, and all are difficult to search and to load completely into a digitized form. This compilation uses results of previous reviews, though many recommendations are updated to incorporate new or omitted data or address errors, and includes recommendations on many reactions that have not been reviewed previously. The database, which incorporates over 50 years of measurements, consists of a total of 2765 recommended bimolecular rate coefficients for the reactions of 1357 organic substances with OH, 709 with Cl, 310 with O3, and 389 with NO3, and is much larger than previous compilations. Many compound types are present in this database, including naturally occurring chemicals formed in or emitted to the atmosphere and anthropogenic compounds such as halocarbons and their degradation products. Recommendations are made for rate coefficients at 298 K and, where possible, the temperature dependences over the entire range of the available data. The primary motivation behind this project has been to provide a large and thoroughly evaluated training dataset for the development of structure–activity relationships (SARs), whose reliability depends fundamentally upon the availability of high-quality experimental data. However, there are other potential applications of this work, such as research related to atmospheric lifetimes and fates of organic compounds, or modelling gas-phase reactions of organics in various environments. This database is freely accessible at https://doi.org/10.25326/36 (McGillen et al., 2019).
- Article
(2106 KB) - Full-text XML
-
Supplement
(527 KB) - BibTeX
- EndNote





The composition of realistic atmospheric and combustion chemical mixtures can be forbiddingly complex, as has recently been emphasized by the advent of automated mechanism generation software (Aumont et al., 2005; Battin-Leclerc et al., 2011; Carter, 2017; Gao et al., 2016). Such complexity presents a major challenge for chemical modellers, since the physical and chemical properties of the vast majority of oxidation products of volatile organic compounds (VOCs) have not been determined experimentally. For example, in the GECKO-A model, the number of possible products formed from a single VOC of only intermediate complexity, α-pinene, can result in ∼ 400 000 different species (Valorso et al., 2011) after a mechanism reduction protocol has been applied. To model the impact of these chemicals on air quality and climate change, information about their chemical and physical behaviour needs to be available. Given the time, expense, and difficulty of making laboratory measurements, it is clear that with current technologies it will be necessary to estimate or compute the properties of almost all of these compounds. To help address this challenge, an expert panel on structure–activity relationship (SAR) evaluation was formed in 2017. This panel has identified several current challenges in atmospheric chemical modelling, which are described in Vereecken et al. (2018). High among the priorities of this group is the assessment of structure–activity relationships for predicting the atmospheric reactivity of VOCs. In this regard, to test the performance of a SAR, it is necessary to compare estimated reaction rate coefficients with available experimental data. The compilation of such data is thus an essential first step in this process, and is the focus of the current work.
Compendia of kinetic data already exist. Notable among these are the thoroughly evaluated datasets provided by the IUPAC Task Group on Atmospheric Chemical Kinetic Data Evaluation (Ammann et al., 2013; Atkinson et al., 2004, 2006, 2007, 2008; Crowley et al., 2010, 2013; IUPAC, 2019), the NASA Panel for Data Evaluation (Burkholder et al., 2015), and the Calvert et al. reviews (Calvert et al., 2000, 2002, 2008, 2011, 2015). The JPL and IUPAC panels are a vital resource, providing detailed evaluations of the major inorganic and organic reactions of importance in atmospheric chemistry, and in fact their reviews of VOC oxidation rate coefficient data (although limited in scope) provide a starting point for our compilation. The work conducted here should be viewed as complementary to these activities, most closely aligned in scope with the Calvert et al. set of reviews. The NIST Chemical Kinetics Database is an extensive compilation of kinetic data (Manion et al., 2015), which, although it is not evaluated, possesses an extremely large scope. Despite the many useful aspects of these resources, they have several drawbacks, such as the following:
-
The frequency with which kinetic data are published is much faster than that with which these data are compiled and reviewed. In the case of IUPAC and JPL, review cycles tend to be ∼ 3–4 years.
-
The number of reaction rates that have been evaluated is considerably smaller than the number that have been determined – in the case of IUPAC and JPL, vastly so. It is therefore inevitable that such evaluations do not currently capture the full chemical diversity available in the experimental literature.
-
The data within these reviews are currently not downloadable in a digital, searchable format.
-
Non-downloadable databases that cannot be accessed offline are subject to downtime (e.g. NIST was recently out of commission for 38 d as a consequence of the 2018–2019 US federal government shutdown). Also, changes can be made to such databases which are not necessarily traceable.
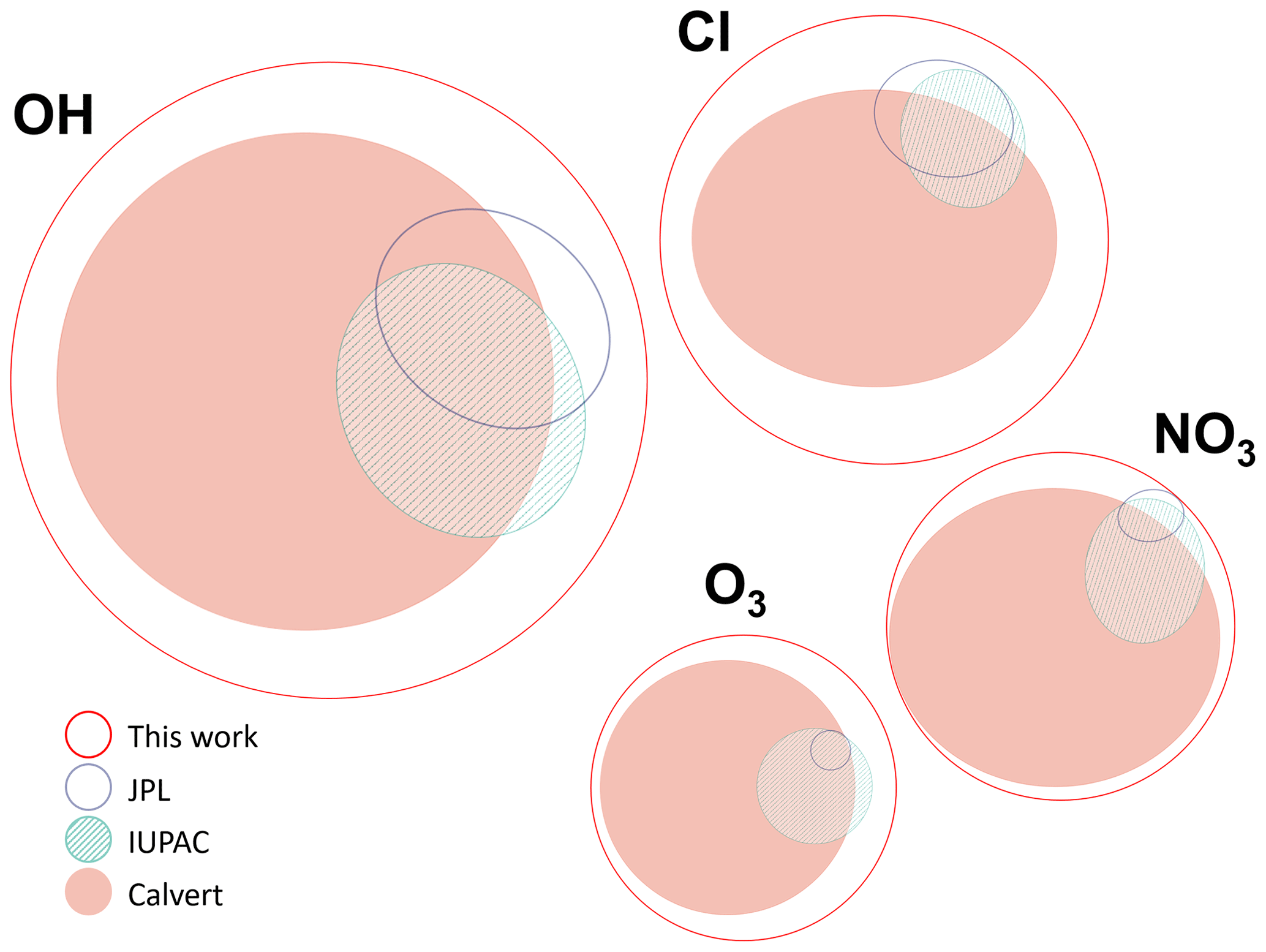
Figure 1Area-proportional Venn diagrams constructed using the eulerAPE software (Micallef and Rodgers, 2014). The size and position of each curve is proportional to the number of species studied with respect to each oxidant, the number of compounds available in each review, and the overlap between these reviews.
The database described here (McGillen et al., 2019) aims to overcome these drawbacks by accounting for the information contained within these previous evaluations and, simultaneously, to augment them by making a new and thorough survey of the chemical kinetics literature. As shown in Fig. 1, the size of the database is considerably larger than previous evaluation projects, which is a consequence of merging each of the available evaluations, new measurements becoming available, and the inclusion of measurements that were overlooked previously.
In addition to this increase in scope, we plan to periodically update this database as new data become available. This is being carried out as part of the activities of the abovementioned SAR evaluation panel (Vereecken et al., 2018). It is intended that this new database will be more agile than these previous efforts, and it will adopt the Earth System Science Data “living data” approach to incorporate new kinetic data that become available and to improve the treatment and description of data herein where necessary. The database can be downloaded in its entirety in the form of an Excel spreadsheet. Our goal is to provide a comprehensive database to serve both as a useful reference source for the kinetics community and as a sound basis upon which to develop SARs for use in atmospheric chemistry and other models.
The reactions of oxidants with organic compounds considered in this compilation can be either bimolecular or termolecular. Bimolecular reactions involve the interaction of two molecules, or an atom and a molecule in the case of the chlorine atom reactions. In termolecular reactions, an excited intermediate is formed, which can be stabilized by collisions with a third body; otherwise decomposition may occur, re-forming reactants. In practice, almost all of the reactions in this compilation are at their high-pressure limit within the pressure and temperature range of interest to atmospheric chemistry, and these reaction rates are therefore readily described as bimolecular reactions.
The rate of a bimolecular chemical reaction is defined in terms of the rate of change of the concentration of reactants or products. The rate coefficient (sometimes referred to as a rate constant) is denoted by the symbol “k” and is the constant of proportionality relating the rate of the reaction to the concentration of the reactants. As an example, for the unit stoichiometry reaction , the rate of reaction is the rate of loss of OH radicals or CH4, or the rate of formation of CH3 radicals or H2O according to Eq. (1):
The units of bimolecular rate coefficients in this database are cm3 molecule−1 s−1, which are those preferred by the atmospheric chemical kinetics community (e.g. Finlayson-Pitts and Pitts, 2000; Seinfeld and Pandis, 2016). Note that “molecule” is not a unit but is typically included for clarity. Where rate coefficients have been reported in units of dm3 mol−1 s−1, these have been multiplied by to convert to units of cm3 molecule−1 s−1.
Rate coefficients are measured using either absolute or relative-rate methods. In absolute measurements, the rate coefficient is determined directly by monitoring the change in concentration of, at least, one of the reactants as a function of time. Typically, experiments are conducted where the pseudo-first-order decay of one reactant is measured under conditions where the other reactant is in excess, such that the concentration of the excess reactant does not change appreciably over time. As an example, flash photolysis can be used to generate OH radicals in the presence of a large excess of CH4. The pseudo-first-order loss of OH can be probed using a variety of techniques such as resonance fluorescence, resonance absorption, or laser-induced fluorescence. The pseudo-first-order loss rate, , is related to the bimolecular rate coefficient k by the expression . Experiments are conducted using different CH4 concentrations, and a plot of k′ versus [CH4] has a slope equal to k.
In relative-rate studies, the rate of the reaction of interest is measured relative to that of a reference reaction whose rate coefficient has been placed on the absolute scale. Having established the rate coefficient of OH+CH4 by an absolute method, this reaction can be used as a reference to measure the rate coefficients for reactions of OH radicals with other organic compounds. As an example, the reactions of OH radicals with a VOC can be studied by exposing mixtures containing the VOC and a reference compound to OH radicals. The reactant and reference compounds are monitored using one or more of the many chromatographic, spectroscopic, and mass spectrometric techniques that are available. A plot of ln ([VOC]t0∕[VOC]) versus ln ([reference]t0∕[reference]t) has a slope equal to the rate coefficient ratio kVOC∕kreference, where t0 and t refer to initial concentrations and concentrations at time t respectively. This plot should be linear and intercept the origin, indicating that secondary chemistry is not significantly affecting the concentrations of the VOC or the reference compound.
In absolute studies, the reaction time must be measured accurately; otherwise systematic errors will be introduced. Furthermore, careful attention must be paid to the reactant purity, where, depending on the relative reactivity of an impurity, even a small fraction (< 0.001) in a sample can affect the retrieved rate coefficient adversely. In some systems, the absolute method is sensitive to regeneration of reactants (e.g. OH recycling), and it is necessary to perform tests to establish that this is not affecting the phenomenological rate coefficient. Conversely, in relative studies, conditions must be selected such that the reactant and reference are lost only by the reaction of interest and that neither reactant nor reference is re-formed in any process. One of the difficulties associated with many relative-rate studies is that they are conducted under chamber conditions, where reactive intermediates from either the VOC of interest or the reference may be present. Therefore, it is often desirable to conduct several measurements in the presence of different reference compounds. It is also better that reaction rates between reference compounds and compounds of interest not be too dissimilar, such that a sufficient amount of chemical conversion is achieved for each in an overlapping timeframe. Absolute-rate techniques are generally capable of higher accuracy than relative-rate methods, once the uncertainties in the reference reaction are considered. Relative-rate techniques are generally simpler to implement and capable of higher precision than absolute-rate techniques. Absolute- and relative-rate methods are complementary and have been used together to provide the wealth of kinetic information documented in this compilation.
The temperature dependence of bimolecular reactions over limited temperature ranges can usually be described by the Arrhenius equation, Eq. (2):
where , the ratio of the activation energy to the gas constant.
The pre-exponential A factor represents the rate of molecular collisions with the correct orientation for reaction, and the exponential term is the fraction of those collisions with sufficient energy for reaction to occur. Over extended temperature ranges many reactions exhibit curved Arrhenius plots because of the importance of multiple reaction pathways each with different temperature dependencies, formation of pre-reactive complexes, and quantum tunnelling at low temperatures, among several other reasons why curvature is expected (Gardiner, 1977). Where rate coefficients have been determined over large temperature ranges and curvature has been observed in Arrhenius space, we have expressed temperature dependences using a three-parameter equation that is sometimes referred to as Kooij's equation (Laidler, 1984) and is referred to here as the “extended” Arrhenius expression, shown in Eq. (3):
Here, an additional term, (T∕300)n, is added to account for the curvature, where “n” is an additional parameter adjusted to fit the data, along with “A” and “B”. Note that this is the same as the standard Arrhenius expression when n=0. We use the (T∕300)n parameterization rather than the simpler Tn because this allows “n” to be dimensionless and the units of A to be independent of n, and of comparable magnitude to the A parameter in the standard expression (see Eq. 2). Although this parameterization is arbitrary and n, A, and B do not have any clear physical or chemical meaning (Carvalho-Silva et al., 2019), it works well in fitting kinetic data for most compounds over wide temperature ranges with only one additional parameter. This is shown, for example, in Fig. 2, which gives an Arrhenius plot for rate coefficient measurements for the reaction of OH with dimethyl ether over a temperature range of 195–1470 K. The dashed blue line is the standard Arrhenius expression derived from the data for 230–300 K, cm3 molecule−1 s−1, while the solid line shows the extended expression, cm3 molecule−1 s−1, which fits the data over the full range of 195–1470 K.
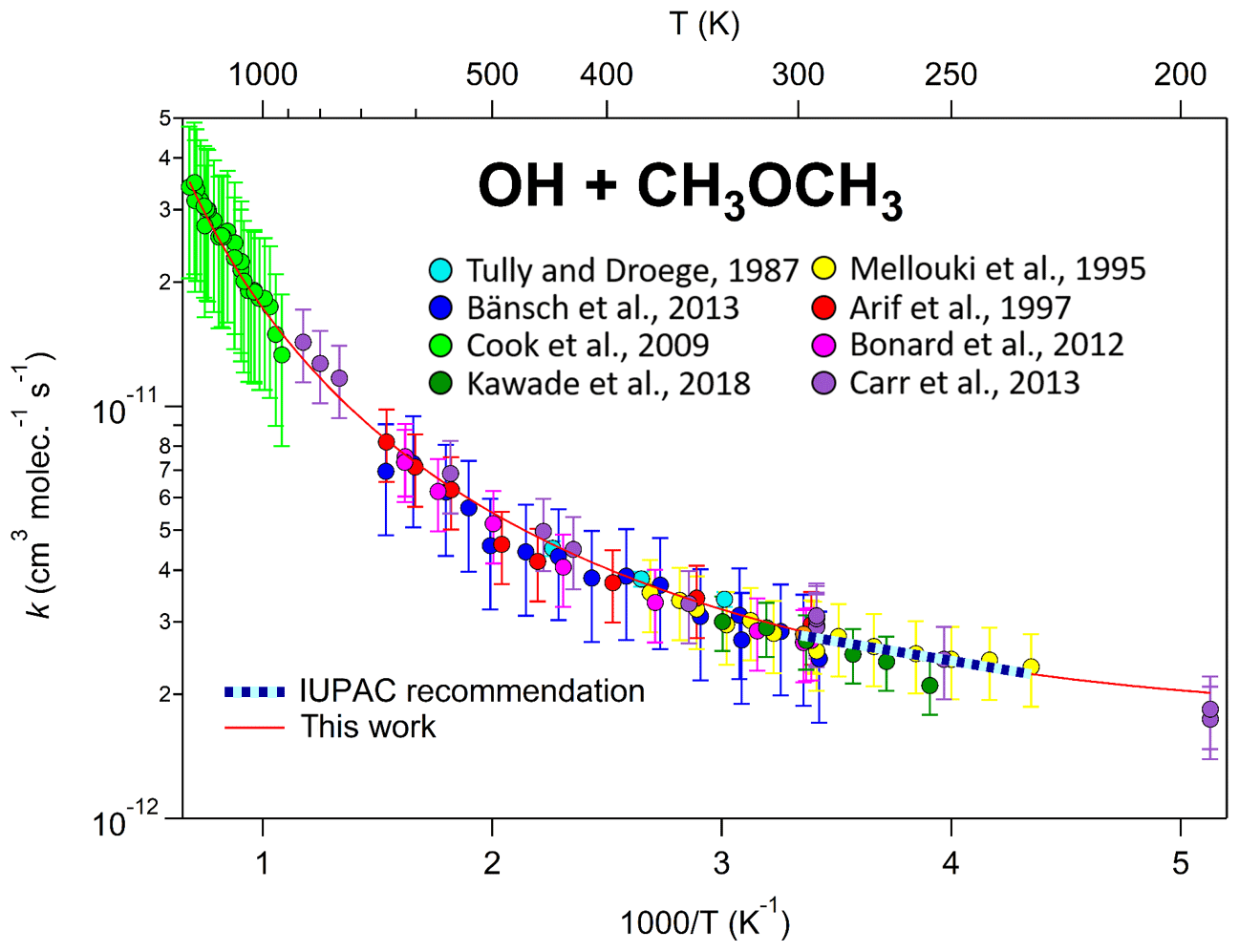
Figure 2Temperature dependence of the reaction of OH with dimethyl ether. As with many other reactions, curvature in Arrhenius space is observed over sufficiently large temperature ranges, especially in systems where quantum tunnelling, pre-reactive complexes, and multiple reaction channels are active. This highlights the need to use the modified Arrhenius expression for some of the reactions in this database.
Whether the measurement is absolute or relative, the vast majority of kinetic studies of organic compounds measure the coefficient for the total reaction, based on the rate of consumption of at least one of the reactants or sometimes from the time-resolved analysis of products. While site-specific information – determined from the quantification of product(s) formed – is available in some cases, these data are not included here. In principle, any rate coefficient that contains several non-degenerate reactive sites can be expressed in the following form, for i number of reactive sites:
It follows that, where data on the branching ratios between these reactive sites are absent, the kinetic information encoded within the total rate coefficient is incomplete. Unfortunately, this is the general state of affairs for the vast majority of reactions contained within the literature, and hence no attempt is made within the framework of the current version of the database to describe branching ratios.
There are approximately 800 reactions in the current database for which the Arrhenius equation has been used to describe the temperature dependence. In the majority of cases, temperature-dependent parameters were taken from previous recommendations, but temperature dependence was re-fitted where problems were identified, such as when temperature ranges were truncated to enable the simpler Arrhenius equation to be used, or where not all data had been incorporated into the recommendation. Data were re-fitted using the extended Arrhenius equation in ∼ 50 cases, all of which were OH reactions. In total, there were 1951 rate coefficients for which only a room temperature rate coefficient was recommended. In the large majority of cases, this was because of an absence of data outside of room temperature. However, in some cases, such as where room temperature determinations were in agreement but where temperature dependences were inconsistent, only the room temperature rate coefficient was recommended. The current database does not contain rate coefficients at other temperatures, other than what could be computed by our recommended temperature-dependent expressions within the stated temperature range.
Reactions occurring on essentially every collision have a rate coefficient known as the gas kinetic limit, which is approximately cm3 molecule−1 s−1 at 298 K, although its precise value will vary with the structure of the reactants. The recommended rate coefficients at 298 K in the present compilation span the range from the gas kinetic limit for the reactions of chlorine atoms with several species to less than approximately 10−22 cm3 molecule−1 s−1 for the reaction of ozone with halogenated alkenes, which corresponds to reaction in approximately 1 out of 1011–1012 collisions. We find that several laboratories have reported rate coefficients (mostly involving reactions with atomic chlorine) that are considerably larger than would be expected from a simple collision theory calculation. For reactants with large dipole moments such as Criegee intermediates, rate coefficients in excess of the collision limit have been rationalized (Chhantyal-Pun et al., 2017, 2018). However, it is more difficult to explain such high rate coefficients in the chlorine reactions, and it is possible that further measurements and theoretical work may be helpful in this regard.
The reviews of IUPAC, JPL, and Calvert et al. (Ammann et al., 2013; Atkinson et al., 2004, 2006, 2007; Burkholder et al., 2015; Calvert et al., 2000, 2002, 2008, 2011, 2015; Crowley et al., 2010, 2013; IUPAC, 2019) constituted the starting point of our data compilation effort. Each recommendation provided in these reviews was transcribed into our own database. Where overlap existed between reviews, recommendations were generally consistent, which provided an opportunity to check for errors in transcription or errors in the reviews. Errors identified in published reviews were excluded from our dataset. Following this initial phase of compilation, kinetic data published between 2015 and present were compiled by searching keywords in Google Scholar over these years and transcribing data from the original publications. Whereas any data published after 2015 cannot be contained in JPL Evaluation Number 18 (Burkholder et al., 2015) and Calvert et al. (2015), IUPAC can be more up to date owing to the more localized update cycle of this review body. The NIST kinetic database was also interrogated to find data that are contained within their extensive database and absent from the reviews that we considered, although, at the time of writing, it is noted that this database has also received no updates since 2015. Following this review of available literature, further, more general searches of kinetic publications were made for all years, which would be able to locate data that had been overlooked by the extensive reviews of Calvert et al. or the large NIST database.
Once all data known to this study were compiled, reviews for individual reactions were made. There are several possible outcomes from entering data into the database, and these are described in Fig. 3.
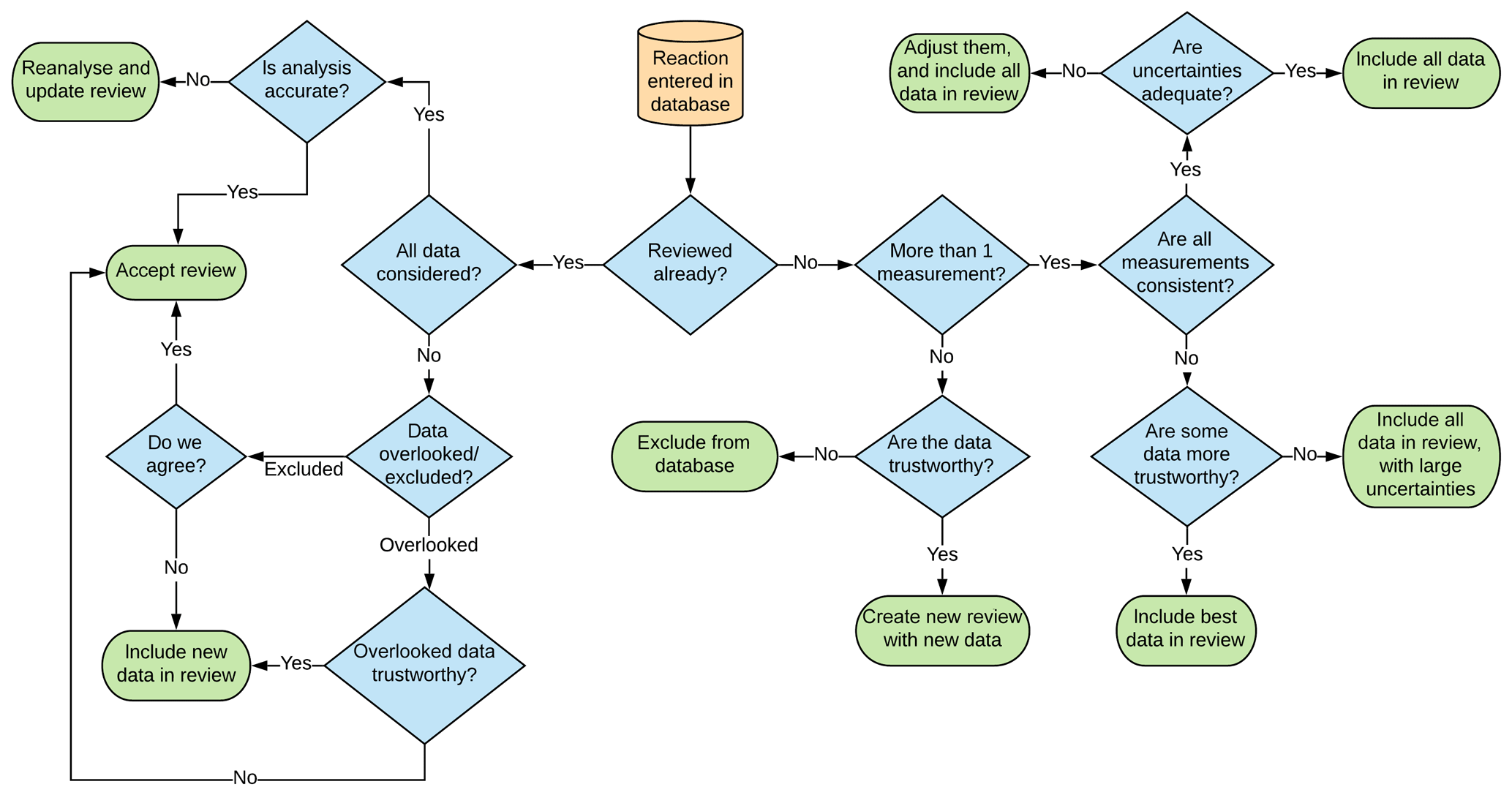
Some of the decisions in this review process are easy to arrive at objectively, such as whether or not all measurements are consistent, which can be determined by a simple comparison. However, other decisions are more nuanced, such as whether or not a measurement is trustworthy. In this instance many factors can influence this decision, including the following:
-
Is the measurement technically difficult?
-
How well was the measurement performed?
-
Were appropriate tests made?
-
Is the apparatus suited to measuring this reaction?
-
Is the measurement generally consistent with analogous reactions?
Because each of these questions requires considerable experience and judgement to answer, the review process was conducted in duplicate and occasionally triplicate, such that, if discrepancies between individual reviewers emerged, these discrepancies were discussed and resolved prior to a final review being accepted by the panel.
Since performing detailed evaluations for each reaction in a database of this size is time-consuming, a streamlined approach to the review process was taken, where a reviewer assessed a longlist of rate coefficients and accepted, rejected, or proposed changes to existing values in the unevaluated database. These actions were compared between reviewers, and, where there was unanimous agreement, values were accepted into the database without further consideration. Subsequently, a shortlist of entries was made, where disagreements were encountered. These were then discussed on an individual basis until a resolution had been reached. Although we consider this approach appropriate for our objective of compiling as comprehensive a compilation of evaluated data as possible within the available amount of time and resources, a number of individual reactions are discussed in more detail in the IUPAC, NASA, and Calvert evaluations, and these are noted in our database as reviews where additional information can be obtained. We therefore consider this work to be complementary to these previous efforts, and, where detailed evaluations exist, readers are directed to the data sheets/notes found within such publications.
Where temperature dependence was available for a reaction, the review process can require that further decisions be made. Firstly, if temperature dependence was determined in some measurements but not others, then A factors were normalized to all available data and the measured Arrhenius temperature dependence parameter, B, was taken from an individual study, or an average of several studies if more than one determination was available. In some instances, where general agreement was observed in A factors but major differences in activation energies were reported, we chose not to recommend temperature-dependent parameters. Secondly, where there are several temperature-dependent studies that span a large range in temperature, and where the temperature dependence can be described adequately by the extended Arrhenius equation (see Eq. 3), an error-weighted linear least squares fit was performed on the entire dataset, and the resultant expression constitutes our recommendation, as shown in Fig. 2, for example. Finally, if temperature dependence information is available but all data are at temperatures higher than 298 K, then extrapolation is necessary to estimate the rate coefficient at 298 K. If the extrapolation is sufficiently close, i.e. causes the rate coefficient to change by less than a factor of 2 compared to that calculated for the lowest temperature in the measurement range, then the extrapolated 298 K rate coefficient is recommended but with an increased uncertainty assignment. We give no 298 K rate coefficient recommendation if the change is greater than that, though the extrapolated rate coefficient is provided as an estimate in the comments. This approach is pragmatic, and more exhaustive treatments are possible; we therefore list this as one of the items of ongoing work listed in Sect. 6.
The structure of the current database, as well as the information it contains, is summarized in the instruction manual and Supplement that is provided with the database file. The database file is an Excel spreadsheet with tables containing the data and also with worksheets giving information about the database, worksheets, and macros for searching and extracting information from the database. The database itself consists of tables giving information about the compounds used, the kinetic data, the references cited, and a table of compound names and other identifiers. The “Compounds” table gives structural information about the compounds, codes indicating the types of compounds that may be useful for search purposes, and the recommended kinetic parameters for the four types of reactions that are currently considered (OH, NO3, O3, and Cl). Most of the compounds have more than one name or identifier that can be used for search purposes, and those that can be used for this application are given in the “Names DB” table. The kinetics data are given in two tables: the “k-Data” table gives the 298 K rate coefficient and temperature dependence parameters from the various reviews or primary studies, while the “kT-Data” table gives the temperatures and rate coefficients that were used for manual fitting. In both cases, codes giving the references used is provided alongside the kinetic data. The reference citations and (where available) URLs for the various references are given in the “References” table.
Table 1Summary of current contents of the experimental atmospheric rate coefficient database for VOCs.

a Numbers of compounds with rate coefficient entries for this reaction. Note that there may be more than one entry per compound because some compounds have more than one type of functional group. b Each entry represents a different reference or source for a rate coefficient, but with no more than one recommended for assessments or SAR development. c Where f=0, compounds contain no substitutions besides C and H, and contain no higher-order bonds; the only compounds that fit this definition are alkanes and cycloalkanes. Halocarbons which may contain many halogen substitutions are treated as one functional group, f=1, because they tend to behave uniformly, unlike complex multifunctional compounds.
The chemical identifiers used included commonly used names that were taken from the original publications or the NCI database (National Cancer Institute, 2010), and other identifiers such as CAS registry numbers when available. Unique textual identifiers (canonical SMILES, InChI, and InChiKey) were also included, making this database easily searchable, such that kinetic information can be obtained rapidly without knowledge of how the molecule is named within the database. There are many chemical informatics software packages and resources that are available for generating SMILES and InChI codes, both freeware (e.g. ACD/ChemSketch, 2018) and commercial software packages such as ChemDraw and online services such as the NCI/CADD Online SMILES Translator (National Cancer Institute, 2017). Note that SMILES strings are not strictly unique and may be dependent upon the algorithm used in a given software implementation; therefore all SMILES were provided in their canonical form as output using the open-source Open Babel software program (O'Boyle et al., 2011). Furthermore, other user-specified differences to SMILES output can still occur, even in canonical form, an important example being the representation of nitrogen–oxygen bonds, where we chose to always represent these bonds as dative, solely for consistency. SMILES strings can be easily converted to canonical SMILES using this package, and InChI and InChiKey using this and other open-source/proprietary resources. Less specific identifiers – including molecular formulae, molecular mass, and types of compounds and functional groups – are also provided, and these can be used to make broader searches to the database possible.
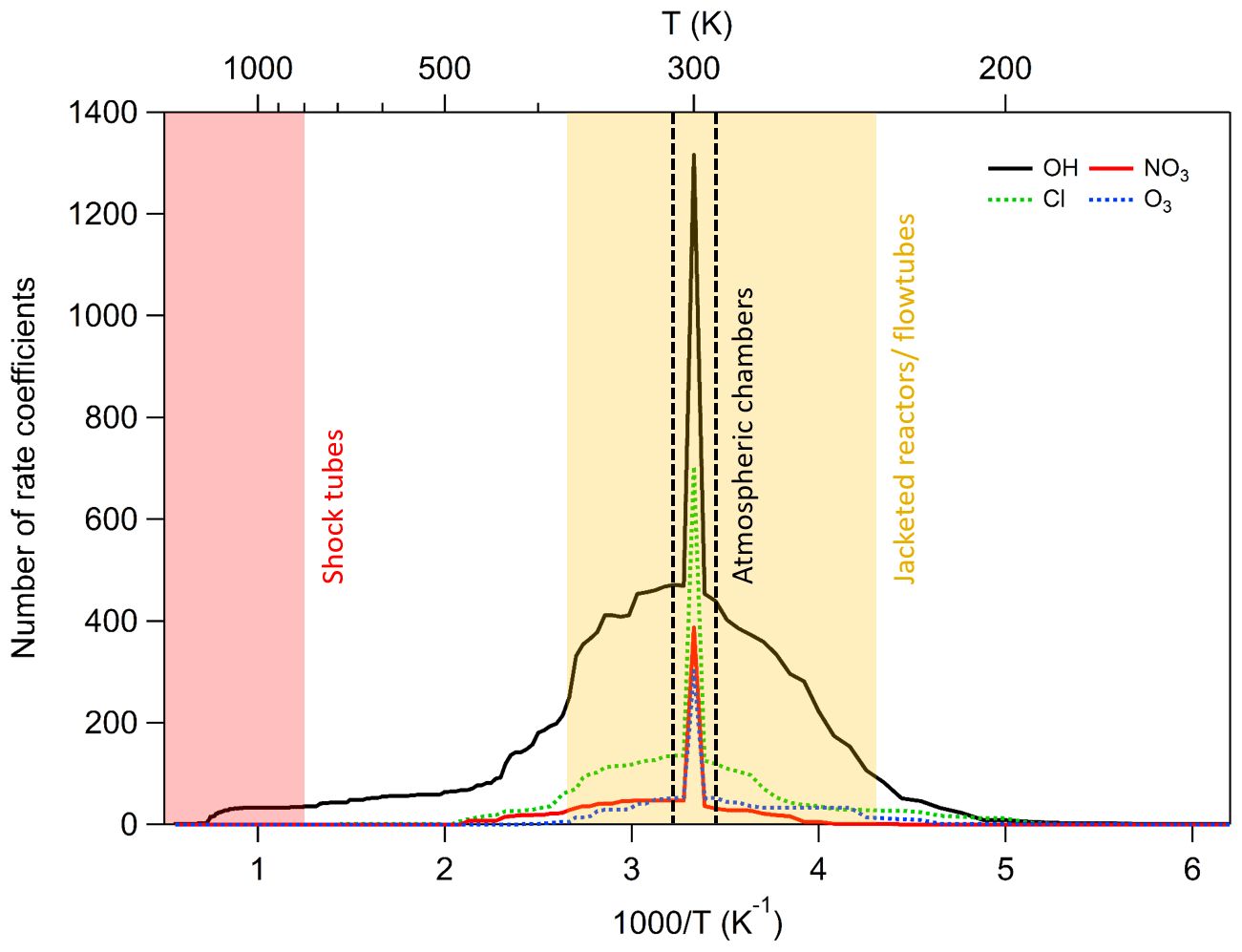
Figure 4Histograms showing the number of rate coefficients at each temperature, separated by oxidant. A large number of reactions have been studied only at room temperature, especially for some oxidants such as NO3, where temperature-dependent data are lacking.
Table 1 is a summary of the number of compounds, reactions, and rate coefficient recommendations in our database, together with the number of non-hydrocarbon functional groups contained within each molecule. As shown above, Fig. 1 provides an overview of the size of the current database in relation to existing compilations of data, and Fig. 4 shows the temperature range covered by our data. From Fig. 4 and Table 1 it is clear that a large majority of data are available at room temperature only or within the range of 250–370 K, which coincides with the general temperature limitations of ambient chamber measurements and jacketed flow reactors respectively. It is also notable that the OH radical dataset possesses the largest number of reactions and the largest fraction of temperature-dependent measurements. By contrast, the number of compounds measured for the NO3 radical is much smaller, and the fraction of temperature-dependent measurements is also much less than for OH.
Regarding the functional form that is used to describe temperature dependences, where obvious curvature can be observed, as shown in Fig. 2 for example, the extended Arrhenius expression is preferred, since this describes the data more faithfully. Ultimately, although most reactions in the OH dataset are expected to be non-Arrhenius, most reactions have yet to be studied over a sufficient temperature range with enough precision to require the third (“n”) parameter of the modified Arrhenius expressions to fit the data, so Arrhenius equations constitute the majority of temperature-dependent expressions in the dataset. By contrast, for reactions such as alkene ozonolysis, where quantum tunnelling is not expected to be feasible, any curvature in Arrhenius space is likely to be small, and so far no ozonolysis reactions known to this study have been shown to exhibit non-Arrhenius behaviour.
As shown in Table 1, of the 1564 compounds studied so far, most reactions have been measured for species that contain two or fewer functional groups. Generally, as the number of functional groups increases in a molecule, the boiling point increases and the saturation vapour pressure decreases, making measurements more challenging in the gas phase, which explains why there are very few measurements on compounds with five or more functional groups. Conversely, for the compounds with no functional groups – defined as a compound that possesses no atom type besides carbon and hydrogen and no higher-order bonds (i.e. alkanes) – the relatively small number of these compounds relates to the fact that there are fewer possible isomers available within the range of volatility that is convenient for experimentation.
As shown in Fig. 1, the database presented in this work is substantially larger than previous compendia and reflects our attempts to compile all available data concerning gas-phase reactions of organic compounds with selected atmospheric oxidants under atmospheric conditions. The current database provides recommendations for the reactions of VOCs with OH and NO3 radicals, O3, and Cl atoms, the major oxidants that react with organic compounds in the atmosphere. Rate coefficients for the reactions of VOCs with other oxidants can be added in later versions of the dataset if there is sufficient interest. However, the focus of the development of the current database is to support the needs of assessing and modelling the impacts of organic compounds in the atmosphere.
For this objective, the ideal is to present rate coefficients for every compound that is emitted into the atmosphere, and for every oxidized organic compound that is formed and reacts in the atmosphere. Knowledge of rate coefficients of emitted compounds is necessary to assess their atmospheric lifetimes and the impacts of their atmospheric reactions on air quality. A total of ∼ 1700 individual compounds have been identified or estimated to be present in the various chemical categories used in US emissions profiles, of which ∼ 1000 compounds are present in VOC mixtures derived to represent total US, California, and Texas anthropogenic emissions (Carter, 2015). This database provides rate coefficient assignments for at least the OH reaction for ∼ 90 % of the mass, though only ∼40 % by number of compounds with non-zero emissions. The high coverage in terms of mass emissions is expected, since such compounds are most likely to be a priority for research. However, a very large number of other species are emitted, and, although individually these may be insignificant, they may become important in the aggregate and should be of interest at least to those who use or emit such compounds. Therefore, it is reasonable to expect that more of these substances will be studied in the future.
Knowledge of the rate coefficients of the oxidized products formed when VOCs react in the atmosphere is necessary for determining the ultimate environmental fates of the emitted compounds and modelling their overall impacts on air quality. However, there are large numbers of possible reactions that many organic compounds and their reactive intermediates can undergo, and the use of automated mechanism generation systems such as GECKO-A (Aumont et al., 2005) is necessary to derive complete mechanisms. Complete coverage of experimental rate coefficients of such a large number of oxidation products is currently unfeasible, and the best that can be hoped for in this regard is to provide rate coefficients for compounds with a variety of representative structures, chemical functionalities, and combinations of functionalities, which may serve as a basis for developing SARs or other methods to estimate rate coefficients for this large array of species.
To obtain an approximate indication of the types of compounds predicted to be formed by mechanism generation systems, and to assess the coverage of this database concerning their rate coefficients, we used GECKO-A to derive complete mechanisms for the atmospheric reactions of the representative compounds n-octane and α-pinene, which are associated with anthropogenic and biogenic activities respectively, and which are expected to yield distinctly different product distributions. The results of this comparison are shown in Figs. 5 and 6.
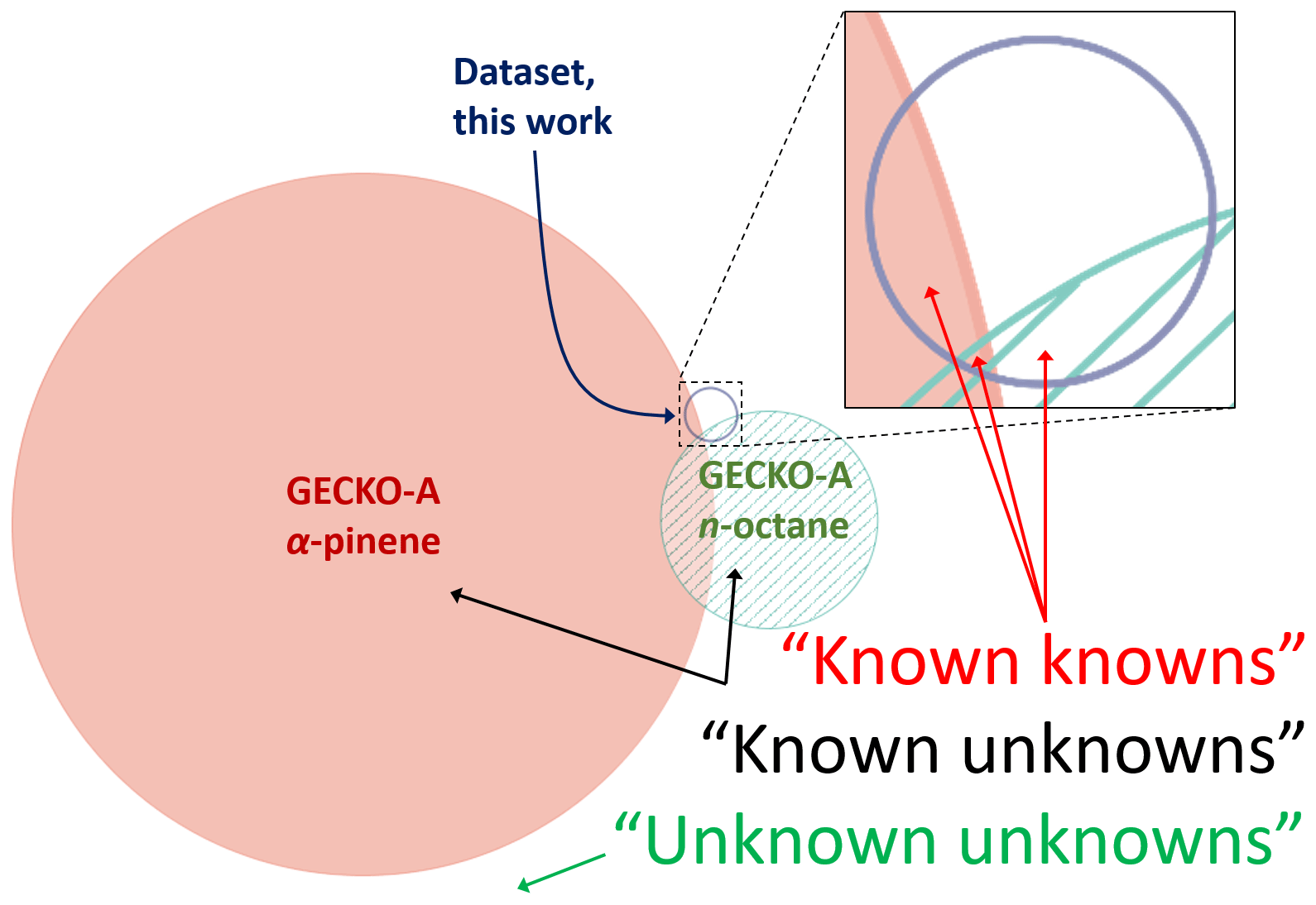
Figure 5Area-proportional Venn diagram showing the overlap between species formed in GECKO-A for n-octane and α-pinene oxidation, and species present in our database. Here, “known knowns” reflect compounds that are formed in GECKO-A for which measurements are available. “Known unknowns” represent chemicals that are formed in GECKO-A for which no measurements are available. “Unknown unknowns” represent species that could be formed from the oxidation of other primary emissions besides n-octane and α-pinene but are not considered in this diagram, and they may also represent compounds that are formed through mechanisms that are currently unknown to/not considered in GECKO-A.
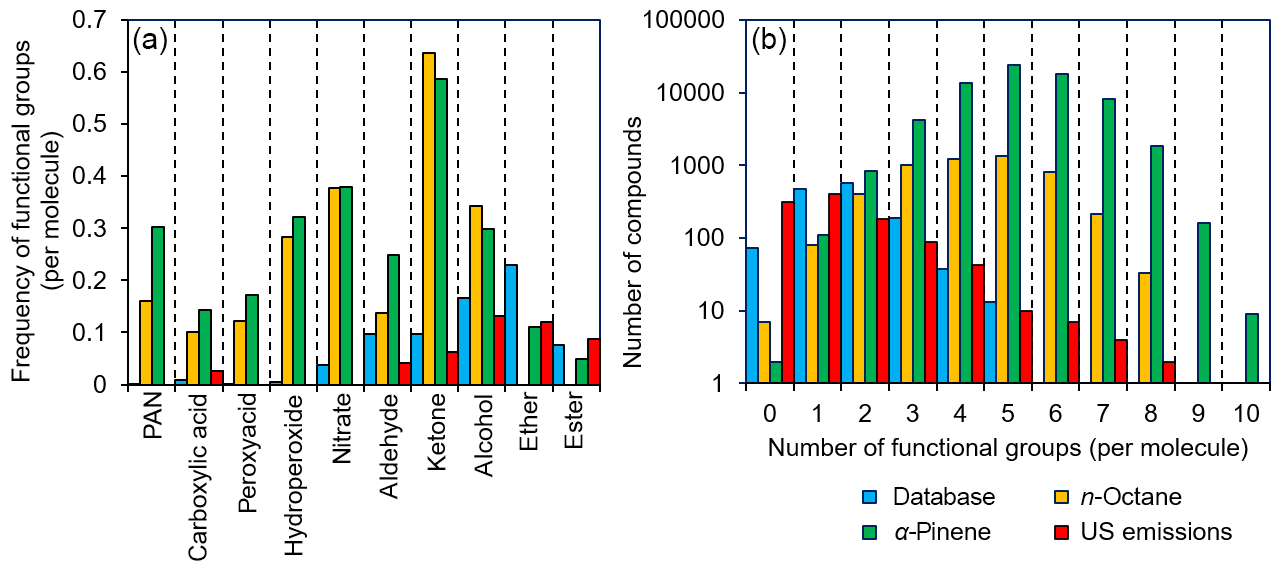
Figure 6Frequency plots comparing the functionalization of compounds within the GECKO-A mechanism of n-octane and α-pinene oxidation, a database of anthropogenic emissions (Carter, 2015), and the compounds present in our kinetic database. (a) Functional groups such as ethers and esters are overrepresented within this database compared with GECKO-A, whereas other functional groups (e.g. nitrates, peroxy acyl nitrates, and hydroperoxides) are very poorly represented. (b) A mismatch is demonstrated between the number of functional groups per molecule in GECKO-A and that of the compounds found in this database. Better agreement is observed in both cases compared with primary emissions profiles.
Figure 5 shows the overlap between the current database and the n-octane and α-pinene products predicted by GECKO-A in terms of individual organic product species (Valorso et al., 2011). The area of each curve in this diagram is proportional to the number of species contained within it. It is clear from this comparison that the number of species that have been studied so far is very small compared with the total number of species produced in the oxidation of these quite structurally simple primary emissions. Furthermore, the overlap between the species studied and the GECKO-A output is vanishingly small. Under the proviso that the GECKO-A mechanism is representative of the state of the knowledge in atmospheric chemistry, species that occupy this overlap region can be regarded as the “known knowns” of atmospheric chemistry, i.e. the species that are known to be produced and have known rate coefficients. When known primary emissions are subjected to the rules of atmospheric chemistry known to GECKO-A, the species that do not overlap with our database are considered as “known unknowns”. The area that falls outside these curves is expected to be vast, and it relates to all species that are formed from all primary emissions that do not overlap with the product distribution of α-pinene, n-octane, or our database. We consider this area to represent “unknown unknowns” in atmospheric chemistry. By this definition, the size of this area cannot be known, but it is anticipated that it is very large, especially when all known and unknown primary emissions are included, and when it is acknowledged that there may be many unusual or exceptional product formation pathways that are currently unknown to the GECKO-A model.
Beyond these three main groupings, there are several other logical criteria by which species that are not contained within chemical mechanisms may be classified. For example, species that are formed through very minor reaction channels may be excluded by simplification protocols that aim to curb the combinatorial explosion within models and may be considered as “unexplored but potentially known unknowns”. Furthermore, for the species which have kinetic measurements but have formation pathways that are currently unknown to chemical mechanisms, these may be considered as “unknown knowns”. These groupings are, however, expected to be small in relation to the unknown unknowns. It is possible that this representation of the state of the knowledge in atmospheric chemistry may be unduly pessimistic, in that these model runs present information on the total number of species but do not account for product fluxes, which could be very small for any species that are produced in rare events.
Notwithstanding, even if the overall flux to the atmosphere was low for a large number of these species, it appears reasonable to expect that given the sheer number of species present in the atmosphere (Goldstein and Galbally, 2007), primarily emitted or produced through oxidation, the fraction of species for which kinetic measurements are available will remain minuscule. This observation is underscored by the fraction of species for which measurements are available over the complete atmospherically relevant temperature range, the bulk of which will be consumed in the troposphere, which experiences temperatures between 220 and 300 K (see Fig. 4). This means that almost all the organic product rate coefficients used by mechanism generation systems like GECKO-A are dependent upon estimation techniques.
From Fig. 6, it is evident that certain functional groups that are relatively uncommon among atmospheric oxidation products of hydrocarbons (e.g. ethers and esters) are well represented in our database, and yet there are many functional groups that are expected to be commonplace that are very much underrepresented (e.g. nitrates, hydroperoxides, peroxyacids, carboxylic acids, and peroxy acyl nitrates). Furthermore, the number of functional groups contained within a molecule is generally smaller in our database (typically between two and three functional groups per molecule) compared with the molecules produced in GECKO-A, where the modal distribution ranges between approximately three and seven functional groups per molecule. The reasons for these disparities are easily rationalized. For example, many of the functional groups that are poorly represented are thermally unstable, and compounds with these functional groups are difficult to purchase, synthesize, store, and handle in experimental studies. Other functional groups, such as the carboxylic acids, are stable, but they suppress vapour pressure to such an extent that only the most volatile members of this family have rate coefficient measurements. Similarly, it is well known that increasing the number of oxygenated functional groups within a molecule reduces the vapour pressure profoundly, and it is therefore often impractical to perform measurements upon highly functionalized species in the gas phase with current technologies and experimental approaches.
As shown in Fig. 6, the situation is more optimistic regarding primary emissions from anthropogenic sources, where industrially important compounds such as ethers, esters, and alcohols are reasonably well represented. Furthermore, the distribution of the number of functional groups per molecule also suggests good overlap. However, it is generally the case that oxidation in the atmosphere will be the predominant fate of each of these primary emissions, and such oxidation will lead to further functionalization. Therefore, as with the example of n-octane and α-pinene oxidation in GECKO-A, it is expected that these primary emissions will generate an immense number of oxidation products under atmospheric conditions.

Figure 7Plots showing correlations between the reaction rates of the various oxidants within this database. Since it is possible that a compound may belong to several of the groupings shown in the legend, categorization of these compounds has been prioritized by reactivity (e.g. an alkene that is also an oxygenate is described as an alkene, since this is likely to be the dominant reactive site).
With such a large number of unknown rate coefficients, it is vital that accurate and computationally inexpensive methods, such as SARs, for estimating rate coefficients are available so that explicit models such as GECKO-A can be employed to make accurate representations of atmospheric chemistry. Although it is anticipated that in-depth analyses of SAR performance will be forthcoming from our expert panel in the future, one well-established method of estimating rate coefficients that arises naturally from the compilation of data presented in our database is that of the correlations exhibited by rate coefficients of VOCs between different oxidants. In Fig. 7, several such relationships are presented. It is clear that some of these relationships are stronger than others. For example, the correlations of ozone with both hydroxyl and chlorine are relatively high, which has been observed previously in the case of O3 and OH (McGillen et al., 2011). In this example, the mechanism of all reactions in these relationships is electrophilic addition. Conversely, other relationships within this diagram involve a combination of addition and abstraction reactions (e.g. any correlations between OH, Cl, and NO3). Furthermore, some reactions may be more affected by steric hindrance (e.g. ozonolysis) than others (McGillen et al., 2008). Consequently, several trends arise depending on the relative efficiencies with which an oxidant participates in a given mechanism. Therefore, when taken as a whole, such correlations appear surprisingly scattered, although it is noted that individual subsets of these correlations may have good predictive power, as has been observed in the OH–Cl correlations for halocarbons and ethers for example (Sulbæk Andersen et al., 2005).
The work contained in the present database represents clear progress in terms of its comprehensive coverage, availability, and accuracy, and the fact that it can be downloaded and readily searched. However, limitations remain and the following future improvements can be envisioned:
-
There are several oxidants that are of importance to combustion chemistry, and there are some atmospheric or laboratory conditions that are not currently included, such as O(3P), O(1D), carbonyl oxides, H and Br atoms, and low-temperature OH reactions.
-
Quantitative information on branching ratios for sites of attack is available for certain reactions, which is not yet implemented in the current database.
-
There are at present only a limited amount of metadata based on experimental conditions, but no information on technique/reactor details/pressure/bath gas/reference compounds in relative-rate experiments.
-
There is a wealth of information published on kinetics in the solution phase that is beyond the scope of the current database, which focuses purely on gas-phase reactions.
-
The current approach to extrapolation of rate coefficients using temperature-dependent data outside 298 K is not statistically rigorous. Improvements will require further data analysis such as that outlined in Hites (2017).
-
Similarly, uncertainty estimates that are ≥ 100 % are not physically meaningful. Improvements upon this may require the asymmetrical distribution of errors afforded by the approach of the IUPAC task group. Again, further statistical analyses will be necessary.
The timescales over which such improvements can be made is likely to depend on external factors such as funding, the continued participation of members of the expert panel, and the possible participation of other experts. However, work will continue on several of these aspects in anticipation of future versions of this database.
The current version of this database, together with instructions on how to use it, is freely available at the following DOI: https://doi.org/10.25326/36 (McGillen et al., 2019).
We present a digital, freely available, searchable, and evaluated compilation of chemical kinetic information with a current focus on gas-phase bimolecular reactions. This database responds to a need within the atmospheric chemistry community and elsewhere for an up-to-date, reviewed database that captures the chemical diversity that is found within the kinetics literature. It is intended that this will be a valuable resource for research into SARs, among other applications, where the quality of training sets will impact accuracy and predictiveness directly. Experimentalists will also be able to use this database to compare their measurements with previous data and analogous compounds, and will also be able to easily locate evaluated reference rate coefficients. Although the current version of this database is the largest database of its kind, there remain many kinetic data that are currently not included in this project, including reactions with several important oxidants, reaction branching ratios, and reactions in other phases besides the gas phase. This, together with the fact that new rate coefficients are published each year, means that further work will be necessary to improve, extend, and maintain this database.
The supplement related to this article is available online at: https://doi.org/10.5194/essd-12-1203-2020-supplement.
All authors contributed to the data compilation, reviewing of data, manuscript writing, and ideas behind the work. Furthermore, WPLC assisted in managing the database, writing Excel macros, and managing the project.
The authors declare that they have no conflict of interest.
Partial support for this project was provided by the Coordinating Research Council (CRC) through contract A-108. However, most of the contributors' efforts in this project were either voluntary or funded by their own projects or institutions. Max R. McGillen thanks Le Studium for their support over part of this project. William P. L. Carter thanks the CRC contract and also the University of California Retirement System for support throughout this project. Abdelwahid Mellouki was supported by the Centre national de la recherche scientifique and also by the Labex Voltaire (ANR-10-LABX-100-01) and the European Union's Horizon 2020 research and innovation programme through the EUROCHAMP-2020 Infrastructure Activity under grant agreement no. 730997. John J. Orlando was supported by the National Center for Atmospheric Research, which is operated by the University Corporation for Atmospheric Research under the sponsorship of the National Science Foundation. The authors also thank AERIS-CNRS and EUROCHAMP-2020 Infrastructure Activity for hosting the database on the EUROCHAMP data centre website.
This research has been supported by the Coordinating Research Council (grant no. contract A-108).
This paper was edited by Vinayak Sinha and reviewed by four anonymous referees.
ACD/ChemSketch: ACD/ChemSketch, Advanced Chemistry Development, Toronto, ON, Canada, 2018.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Arif, M., Dellinger, B., and Taylor, P. H.: Rate Coefficients of Hydroxyl Radical Reaction with Dimethyl Ether and Methyl tert-Butyl Ether over an Extended Temperature Range, J. Phys. Chem. A, 101, 2436–2441, https://doi.org/10.1021/jp963119w, 1997.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625-4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species, Atmos. Chem. Phys., 8, 4141–4496, https://doi.org/10.5194/acp-8-4141-2008, 2008.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Bänsch, C., Kiecherer, J., Szöri, M., and Olzmann, M.: Reaction of Dimethyl Ether with Hydroxyl Radicals: Kinetic Isotope Effect and Prereactive Complex Formation, J. Phys. Chem. A, 117, 8343–8351, https://doi.org/10.1021/jp405724a, 2013.
Battin-Leclerc, F., Blurock, E., Bounaceur, R., Fournet, R., Glaude, P.-A., Herbinet, O., Sirjean, B., and Warth, V.: Towards cleaner combustion engines through groundbreaking detailed chemical kinetic models, Chem. Soc. Rev., 40, 4762–4782, https://doi.org/10.1039/c0cs00207k, 2011.
Bonard, A., Daële, V., Delfau, J.-L., and Vovelle, C.: Kinetics of OH Radical Reactions with Methane in the Temperature Range 295–660 K and with Dimethyl Ether and Methyl-tert-butyl Ether in the Temperature Range 295–618 K, J. Phys. Chem. A, 106, 4384–4389, https://doi.org/10.1021/jp012425t, 2002.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies, evaluation no. 18, available at: http://jpldataeval.jpl.nasa.gov (last access: 25 May 2020), 2015.
Calvert, J., Atkinson, R., Kerr, J., Madronich, S., Moortgat, G., Wallington, T., and Yarwood, G.: The Mechanisms of Atmospheric Oxidation of Alkenes, Oxford University Press, 2000.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. J., and Yarwood, G.: The mechanisms of atmospheric oxidation of aromatic hydrocarbons, Oxford University Press, Oxford, New York, 2002.
Calvert, J. G., Derwent, R. G., Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: Mechanisms of the atmospheric oxidations of the alkanes, Oxford University Press, 2008.
Calvert, J. G., Mellouki, A., Orlando, J. J., Pilling, M. J., and Wallington, T. J. (Eds.): The mechanisms of atmospheric oxidation of the oxygenates, Oxford Univ. Press, Oxford, 2011.
Calvert, J. G., Orlando, J. J., Stockwell, W. R., and Wallington, T. J.: The mechanisms of reactions influencing atmospheric ozone, Oxford University Press, Oxford New York Aukland, 2015.
Carr, S. A., Still, T. J., Blitz, M. A., Eskola, A. J., Pilling, M. J., Seakins, P. W., Shannon, R. J., Wang, B., and Robertson, S. H.: Experimental and Theoretical Study of the Kinetics and Mechanism of the Reaction of OH Radicals with Dimethyl Ether, J. Phys. Chem. A, 117, 11142–11154, https://doi.org/10.1021/jp4070278, 2013.
Carter, W. P. L.: Development of a database for chemical mechanism assignments for volatile organic emissions, JAPCA J. Air Waste Ma., 65, 1171–1184, https://doi.org/10.1080/10962247.2015.1013646, 2015.
Carter, W. P. L.: Gateway to the SAPRC-16 Mechanism Generation System, Gateway to the SAPRC-16 Mechanism Generation System, available at: http://mechgen.cert.ucr.edu/ (last access: 25 May 2020), 2017.
Carvalho-Silva, V. H., Coutinho, N. D., and Aquilanti, V.: Temperature Dependence of Rate Processes Beyond Arrhenius and Eyring: Activation and Transitivity, Front. Chem., 7, 380, https://doi.org/10.3389/fchem.2019.00380, 2019.
Chhantyal-Pun, R., McGillen, M. R., Beames, J. M., Khan, M. A. H., Percival, C. J., Shallcross, D. E., and Orr-Ewing, A. J.: Temperature-Dependence of the Rates of Reaction of Trifluoroacetic Acid with Criegee Intermediates, Angew. Chem. Int. Edit., 56, 9044–9047, https://doi.org/10.1002/anie.201703700, 2017.
Chhantyal-Pun, R., Rotavera, B., McGillen, M. R., Khan, M. A. H., Eskola, A. J., Caravan, R. L., Blacker, L., Tew, D. P., Osborn, D. L., Percival, C. J., Taatjes, C. A., Shallcross, D. E., and Orr-Ewing, A. J.: Criegee Intermediate Reactions with Carboxylic Acids: A Potential Source of Secondary Organic Aerosol in the Atmosphere, ACS Earth and Space Chemistry, 2, 833–842, https://doi.org/10.1021/acsearthspacechem.8b00069, 2018.
Cook, R. D., Davidson, D. F., and Hanson, R. K.: High-Temperature Shock Tube Measurements of Dimethyl Ether Decomposition and the Reaction of Dimethyl Ether with OH, J. Phys. Chem. A, 113, 9974–9980, https://doi.org/10.1021/jp902403n, 2009.
Crowley, J. N., Ammann, M., Cox, R. A., Hynes, R. G., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V – heterogeneous reactions on solid substrates, Atmos. Chem. Phys., 10, 9059–9223, https://doi.org/10.5194/acp-10-9059-2010, 2010.
Crowley, J. N., Ammann, M., Cox, R. A., Hynes, R. G., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Corrigendum to “Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V – heterogeneous reactions on solid substrates” published in Atmos. Chem. Phys. 10, 9059–9223, 2010, Atmos. Chem. Phys., 13, 7359–7359, https://doi.org/10.5194/acp-13-7359-2013, 2013.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the upper and lower atmosphere: theory, experiments, and applications, Academic Press, San Diego, 2000.
Gao, C. W., Allen, J. W., Green, W. H., and West, R. H.: Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms, Comput. Phys. Commun., 203, 212–225, https://doi.org/10.1016/j.cpc.2016.02.013, 2016.
Gardiner, W. C.: Temperature dependence of bimolecular gas reaction rates, Accounts Chem. Res., 10, 326–331, https://doi.org/10.1021/ar50117a003, 1977.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Hites, R. A.: Calculating the Confidence and Prediction Limits of a Rate Constant at a Given Temperature from an Arrhenius Equation Using Excel, J. Chem. Educ., 94, 398–400, https://doi.org/10.1021/acs.jchemed.6b00842, 2017.
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, Task Group on Atmospheric Chemical Kinetic Data Evaluation, available at: http://iupac.pole-ether.fr/ (last access: 25 May 2020), 2019.
Kawade, M., Sharma, A., Srinivas, D., Saha, A., Upadhyaya, H. P., Kumar, A., and Naik, P. D.: Rate coefficients of hydroxyl radical reaction with dimethyl ether over a temperature range of 257–333 K, Chem. Phys. Lett., 706, 558–563, https://doi.org/10.1016/j.cplett.2018.06.065, 2018.
Laidler, K. J.: The development of the Arrhenius equation, J. Chem. Educ., 61, 494–498, https://doi.org/10.1021/ed061p494, 1984.
Manion, J. A., Huie, R. E., Levin, R. D., Burgess Jr., D. R., Orkin, V. L., Tsang, W., McGivern, W. S., Hudgens, J. W., Knyazev, V. D., Atkinson, D. B., Chai, E., Tereza, A. M., Lin, C.-Y., Allison, T. C., Mallard, W. G., Westley, F., Herron, J. T., Hampson, R. F., and Frizzell, D. H.: NIST Chemical Kinetics Database, Standard Reference Database 17, Version 7.0 (Web Version), Release 1.6.8 Data Version 2015.09, A compilation of kinetics data on gas-phase reactions, NIST Chemical Kinetics Database, available at: http://kinetics.nist.gov/ (last access: 25 May 2020), 2015.
McGillen, M. R., Carey, T. J., Archibald, A. T., Wenger, J. C., Shallcross, D. E., and Percival, C. J.: Structure-activity relationship (SAR) for the gas-phase ozonolysis of aliphatic alkenes and dialkenes, Phys. Chem. Chem. Phys., 10, 1757–1768, https://doi.org/10.1039/b715394e, 2008.
McGillen, M. R., Archibald, A. T., Carey, T., Leather, K. E., Shallcross, D. E., Wenger, J. C., and Percival, C. J.: Structure–activity relationship (SAR) for the prediction of gas-phase ozonolysis rate coefficients: an extension towards heteroatomic unsaturated species, Phys. Chem. Chem. Phys., 13, 2842–2849, https://doi.org/10.1039/C0CP01732A, 2011.
McGillen, M. R., Carter, W. P. L., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., and Wallington, T. J.: Database for the Kinetics of the Gas-Phase Atmospheric Reactions of Organic Compounds, Database for the Kinetics of the Gas-Phase Atmospheric Reactions of Organic Compounds, https://doi.org/10.25326/36, 2019.
Mellouki, A., Teton, S., and Le Bras, G.: Kinetics of OH radical reactions with a series of ethers, Int. J. Chem. Kinet., 27, 791–805, https://doi.org/10.1002/kin.550270806, 1995.
Micallef, L. and Rodgers, P.: eulerAPE: Drawing Area-Proportional 3-Venn Diagrams Using Ellipses, edited by: Kestler, H. A., PLoS ONE, 9, e101717, https://doi.org/10.1371/journal.pone.0101717, 2014.
National Cancer Institute: Downloadable Structure Files of NCI Open Database Compounds, Downloadable Structure Files of NCI Open Database Compounds, available at: https://cactus.nci.nih.gov/download/nci/ (last access: 7 August 2019), 2010.
National Cancer Institute: Online SMILES Translator and Structure File Generator, Online SMILES Translator and Structure File Generator, available at: https://cactus.nci.nih.gov/translate/ (last access: 7 August 2019), 2017.
O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R.: Open Babel: An open chemical toolbox, J. Cheminformatics, 3, 33, https://doi.org/10.1186/1758-2946-3-33, 2011.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 3rd edn., John Wiley & Sons, Hoboken, New Jersey, 2016.
Sulbæk Andersen, M. P., Nielsen, O. J., Wallington, T. J., Hurley, M. D., and DeMore, W. B.: Atmospheric Chemistry of CF3OCF2CF2H and CF3OC(CF3)2H: Reaction with Cl Atoms and OH Radicals, Degradation Mechanism, Global Warming Potentials, and Empirical Relationship between k (OH) and k (Cl) for Organic Compounds, J. Phys. Chem. A, 109, 3926–3934, https://doi.org/10.1021/jp044635m, 2005.
Tully, F. P. and Droege, A. T.: Kinetics of the reactions of the hydroxyl radical with dimethyl ether and diethyl ether, Int. J. Chem. Kinet., 19, 251–259, https://doi.org/10.1002/kin.550190308, 1987.
Valorso, R., Aumont, B., Camredon, M., Raventos-Duran, T., Mouchel-Vallon, C., Ng, N. L., Seinfeld, J. H., Lee-Taylor, J., and Madronich, S.: Explicit modelling of SOA formation from α-pinene photooxidation: sensitivity to vapour pressure estimation, Atmos. Chem. Phys., 11, 6895–6910, https://doi.org/10.5194/acp-11-6895-2011, 2011.
Vereecken, L., Aumont, B., Barnes, I., Bozzelli, J. W., Goldman, M. J., Green, W. H., Madronich, S., McGillen, M. R., Mellouki, A., Orlando, J. J., Picquet-Varrault, B., Rickard, A. R., Stockwell, W. R., Wallington, T. J., and Carter, W. P. L.: Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 50, 435–469, https://doi.org/10.1002/kin.21172, 2018.